Os defeitos cardíacos congênitos do coração (DCC) ou anomalias cardíacas congênitas ocorrem em uma proporção de 6 a 8 bebês para cada 1000 nascidos vivos. As principais etiologias dessas anormalidades são de origem genética e exposição a teratógenos, sendo que o período de maior vulnerabilidade aos agentes teratogênicos está provavelmente entre a 2ª e 8ª semanas (antes da cardiogênese visível e o fim do período embrionário, respectivamente). As principais malformações cardíacas consistem em: defeitos no septo interventricular, defeitos no septo interatrial, defeitos nas valvas atrioventriculares, estenose das valvas semilunares, defeitos na septação do trato de saída e Tetralogia de Fallot.
Defeito no septo atrial (DSA)
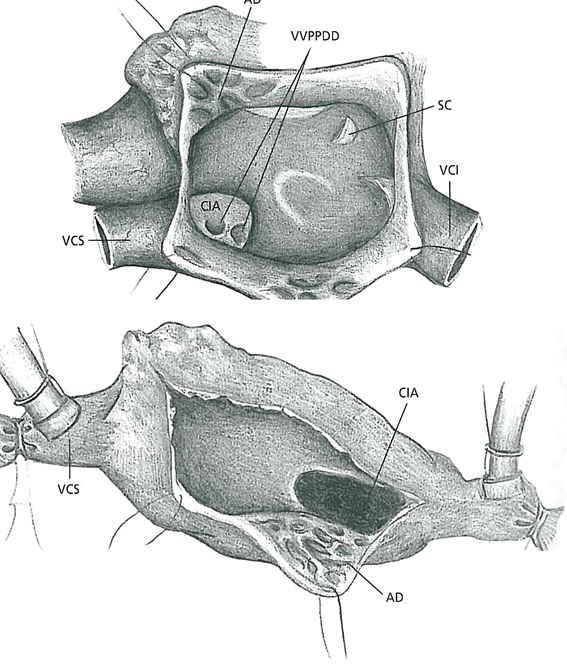
Ocorre quando o septum secundum é muito curto para cobrir o forâmen secundum(ou quando o forâmen secundum é muito largo). As aberturas na porção baixa do septo interatrial são chamadas do tipo forâmen primum e estão fortemente associada a um defeito do canal atrioventricular, frequente na trissomia do 21 (síndrome de down). As aberturas mais altas geralmente se localizam na região da fossa oval e ocorrem por excesso de reabsorção do septum primum, produzindo um grande forâmen secundum.
Dessa forma, os defeitos no septum secundum causam vazamento de sangue do átrio esquerdo para o átrio direito. O aumento do fluxo para as câmaras direitas pode levar ao alargamento de átrio e ventrículo ocasionando, posteriormente, arritmias atriais.. Outro defeito cardíaco, porém, muito raro, é o átrio comum. Neste caso, o septo interatrial está ausente pelo não desenvolvimento do septum primum e secundum.
Defeito no septo ventricular (DSV)
A comunicação entre ventrículos é a anormalidade cardíaca mais comum sendo que o defeito no septo ventricular possui várias causas. São elas: desenvolvimento deficiente das saliências do cone-tronco proximais, falência na fusão do septo ventricular muscular e membranáceo, defeito no septo atrioventricular e desenvolvimento insuficiente do septo muscular interventricular. Geralmente, ocorre um desvio de sangue da esquerda para a direita, podendo ocorrer em grandes DSV, excessivo fluxo sanguíneo pulmonar e hipertensão pulmonar, que levam à dispnéia (dificuldade respiratória) e insuficiência cardíaca na infância. A maioria dos casos se resolve espontaneamente, mas quando necessário, o fechamento pode ser cirúrgico.
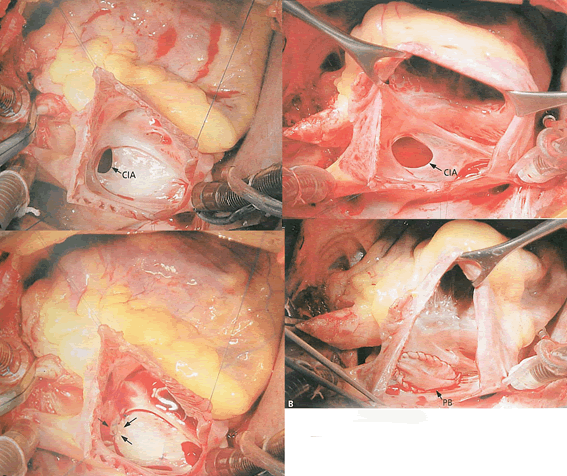
Comunicação interatrial
Fonte: Cardiologia e Cirurgia Cardiovascular Pediátrica, Croti et al., 2008. 1ª edição. Editora Roca.
Defeito nas valvas atrioventriculares
Surgem a partir de erros no remodelamento responsável pela formação dos folhetos das valvas, das cordas tendíneas, dos músculos papilares, do coxim endocárdico e do miocárdio ventricular. A patogênese da atresia das valvas não é compreendida. Na atresia de valva tricúspide, a comunicação entre o átrio direito e ventrículo direito não ocorre. Dessa forma, o sangue que chega ao átrio direito vaza para o átrio esquerdo através do forame oval persistente. Dessa forma, o coração funciona como um coração univentricular, pois a circulação é dirigida somente para o ventrículo esquerdo. Consequentemente, o ventrículo direito torna-se hipoplásico enquanto o ventrículo esquerdo fica alargado. Outra situação em que geralmente as valvas atrioventriculares são anormais é no Ventrículo com Dupla Saída (VDDS), situação na qual as grandes artérias (aórtica e pulmonar) saem totalmente, ou predominantemente, do ventrículo direito.
Ventrículo com dupla saída

Fonte: Cardiologia e Cirurgia Cardiovascular Pediátrica, Croti et al., 2008. 1ª edição. Editora Roca.
Estenose das valvas semilunares
A estenose das valvas semilunares está relacionada à valva aórtica ou à valva pulmonar. Na estenose de valva aórtica ocorre a fusão das bordas das válvulas o que leva a um estreitamento de sua abertura. Esta alteração pode ser adquirida ou congênita, sendo que esta última ocorre por um erro na cavitação e no remodelamento do coxim responsável pela formação da valva semilunar aórtica resultando em uma bi comissura de valva aórtica (formação de apenas dois folhetos, ao invés de três). Como repercussão, a estenose de valva aórtica leva à hipertrofia de ventrículo esquerdo, hipertensão pulmonar e futura falência cardíaca. A estenose da valva pulmonar geralmente está presente com outras alterações como é visto na Tetralogia de Fallot.
Este defeito está relacionado com a falha de formação do septo que divide o tronco arterial em aorta e tronco pulmonar, levando ao tronco arterial persistente (TA). Acredita-se que a causa dessa má-formação é o desenvolvimento anormal das células da crista neural e sempre está associada com um defeito no septo ventricular. Dessa maneira, os bebês que apresentam TA tem seu sangue misturado no trato de saída comum e um vazamento da esquerda para a direita em direção ao lado pulmonar ocasionando em hipertensão pulmonar.
Outro defeito na septação do tronco arterial é quando ela não ocorre de maneira padrão em espiral, levando à transposição dos grandes vasos ou artérias (TGA). Dessa forma, o ventrículo esquerdo se esvazia na circulação pulmonar e o ventrículo direito na circulação sistêmica. A TGA geralmente está associada com outras anomalias cardíacas (DSV, DSA, por exemplo) e com a persistência do ducto arterial. Consequentemente, os defeitos DSA e DSV associados favorecem alguma troca entre as circulações sistêmica e pulmonar. As crianças que apresentam TGA sem correção cirúrgica morrem em poucos meses.
Transposição dos grandes vasos

Fonte: Cardiologia e Cirurgia Cardiovascular Pediátrica, Croti et al., 2008. 1ª edição. Editora Roca.
Tetralogia de Fallot
A Tetralogia de Fallot é uma síndrome que se caracteriza pela presença de quatro malformações clássicas e que ocorrem simultaneamente. São elas: estenose pulmonar, defeito no septo ventricular, deslocamento da aorta para a direita (aorta acavalada) e hipertrofia ventricular direita. Todas essas alterações juntas favorecem o aumento da pressão sanguínea no ventrículo direito resultando em hipertrofia do ventrículo direito.
Bibliografia Consultada:
Moore KL, Persaud TVN. Embriologia clínica. 8a ed. Rio de Janeiro (RJ): Elsevier; 2008.
Moore KL, Persaud TVN. The developing human: clinically oriented embryology. 7th ed. Philadelphia: WB Saunders; 2003.
Schoenwolf GC, Bleyl SB, Brauer PR, Francis-West PH. Larsen embriologia humana. 4a ed. Rio de Janeiro (RJ): Elsevier; 2009.
O'Rahilly R, Müller F. Embriologia & teratologia humanas. 3a ed. Rio de Janeiro (RJ): Guanabara Koogan; 2005.
Croti UA, Mattos SS, Pinto Júnior VC, Aiello V. Cardiologia e cirurgia cardiovascular pediátrica. São Paulo: Roca; 2008.
1) Desenvolvimento do Coração
2) Desenvolvimento das Veias
3) Desenvolvimento das Artérias
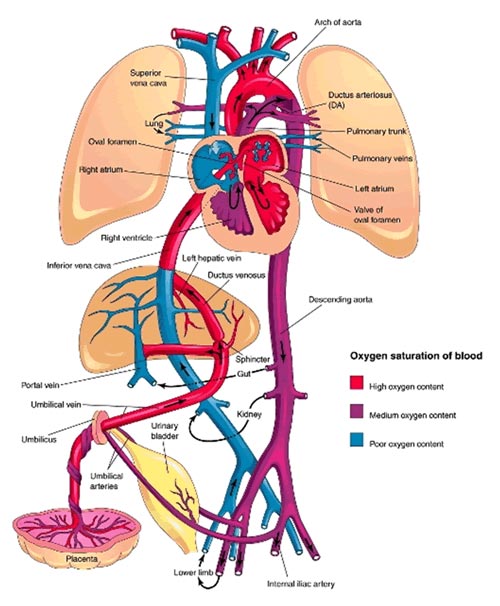
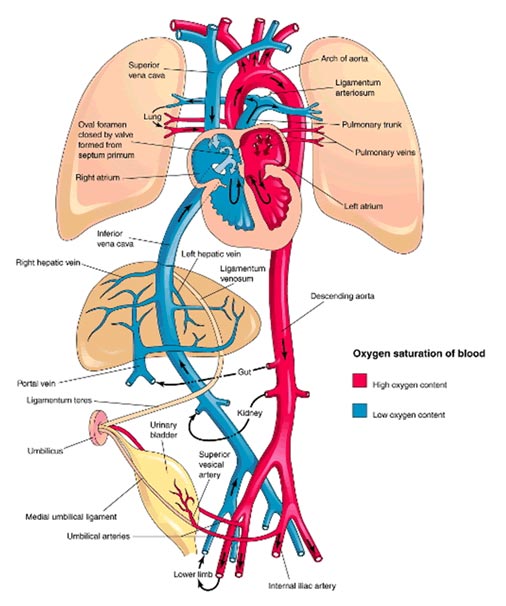
4) Circulação Fetal
5) Anomalias Cardíacas Congênitas |